Understanding Long QT Syndrome Caused by Genetic Variants
by Amanda Chase, PhD
January 24, 2024
Ogden syndrome is an ultra-rare disorder occurring in infancy that is characterized by developmental delay, intellectual disability, failure to thrive, and cardiac anomalies, among other symptoms. Ogden syndrome is caused by a single error (mutation) in a gene called NAA10. Genes have the instructions for making proteins, written out by a sequence of amino acid bases A, T, G, C. While errors in sequence occur often, they are only a problem if occurring on a vital gene in a crucial spot, which can then have devastating consequences. A single mutation in NAA10 is one such example.
Proteins are often considered a “workhorse” of the cell, as they are responsible for an incredible number of tasks. Certain changes or modifications to proteins fine-tune their function, acting as a tag that influences how the protein works. Acetylation is one such modification. NAA10 (N-alpha-acetyltransferase 10) plays a vital role in the acetylation process, making NAA10 essential for the proper function of various biological processes in the human body. A mutation in NAA10 is associated with Ogden syndrome and triggers severe physical manifestations. These genetic changes are carried by both men and women but are more symptomatic in men. The complexity of the clinical manifestations caused by a single mutation highlight the importance of NAA10 protein in multiple organs.
The present study focuses on the cardiac defects observed in Ogden syndrome patients. The heart contracts and relaxes to pump blood in a very coordinated manner that is controlled by electrical signals, referred to as action potential. These signals tell the heart to contract and relax, creating what we know as the heartbeat. There is a period to recharge between each beat. Prolonged QT intervals (Long QT syndrome; LQT) means a longer time than usual to relax which delays the time to recharge. This can lead to life-threatening arrhythmias. In a recent study published in Circulation, Stanford Cardiovascular Institute affiliated first author Nadjet Belbachir, PhD, and senior author Joseph Wu, MD, PhD, used cells from patients with Ogden syndrome to assess the ability to retain the LQT phenotype and to understand the mechanism behind the action potential prolongation.
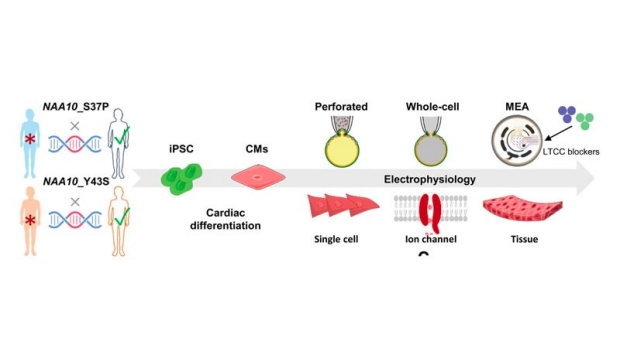
Study Overview. iPSC-CMs were derived from 2 patients carrying mutations on the NAA10 gene. Corresponding CRISPR corrected lines were also created. These iPSC-CM lines were used for a full cellular electrophysiology investigation.
In this study, two patients were recruited who had different NAA10 mutations (variants), one showed more mild symptoms (Y43S) and one had severe symptoms (S37P). Induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) were created from these two patients. To provide a sophisticated manner of studying these variants, CRIPSR was used to create corrected pluripotent stem cell lines. CRISPR allows the researcher to change the written sequence as they want. Here, they made changes so the iPSC-CMs had NAA10 without any mutations, thus called “corrected”.
Action potential recordings were able to confirm that the NAA10 variants showed the LQT phenotype, and the corrected lines did not. This enhances the understanding of the link between NAA10 variants and cardiac arrhythmia, further contributing to the study of NAA10 variant-related dysfunction.
Calcium channels contribute to electrical stability during action potential generation and plateau phase. In the heart, this means playing an important role in regulating cardiac muscle contraction associated with the heartbeat. In this study, the research team demonstrated that the action potential prolongations resulted from changes to a calcium channel (Cav1.2) that led to an increase of ICAL current density, ultimately leading to the LQT phenotype. Intriguingly, providing blockers of ICAL successfully corrected the action potential prolongation in the patient iPSC-CMs. This could suggest the potential for a therapeutic tool for treatment of NAA10 variants resulting in LQT and associated arrhythmia.
Other research team members affiliated with Stanford Cardiovascular Institute are Mengchen Shen, Sophia L. Zhang, Joe Z. Zhang, Chun Liu, and Ning Ma.

Dr. Nadjet Belbachir

Dr. Joseph Wu