Revealing the Path: Pulmonary Arterial Hypertension
by Micaela Harris
February 14, 2024
Pulmonary arterial hypertension (PAH) is a condition in which arteries in the lungs thicken and narrow the channels through which blood flows, thereby raising the pressure against which the heart needs to pump. This can ultimately lead to heart failure.
PAH is often classified into different subtypes based on its cause. Hereditary pulmonary arterial hypertension occurs due to a mutation in a receptor that is on the surface of vascular cells, while idiopathic pulmonary hypertension lacks a clear cause. Regardless of subtypes, there are extremely limited therapeutic treatments for the disease, with a 40% mortality rate over 5 years.
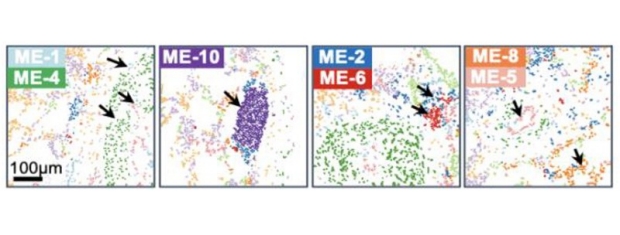
Pulmonary artery cell groupings cluster in local microenvironments (MEs). MEs are: immune and smooth muscle cell-enriched (ME-1), immune-enriched (ME-2), SMC-enriched (ME-4), endothelial-enriched (ME-5), Cd8 T cell-enriched (ME-6), and B cell-enriched (ME-10). Please refer to the paper for the full image.
Researchers at Stanford University, led by Selena Ferrian, PhD, Michael Angelo, MD, PhD, and Marlene Rabinovitch, MD, investigated the immune cell landscape in pulmonary arteries and surrounding lung tissue to unravel the intricate mechanisms underlying the initiation and progression of PAH. Their groundbreaking study, recently published in The American Journal of Respiratory and Critical Care Medicine, sheds light on the multifaceted nature of this disease. By uncovering the cellular intricacies of PAH, their research offers valuable insights into disease progression and paves the way for the development of tailored therapies.
Using state-of-the-art multiplexed ion beam imaging by time-of-flight, a technology leveraging metal-tagged antibodies for precise detection, the team analyzed thirty-five markers within preserved tissue. These markers spanned immune cell differentiation, activation, and tissue-specific markers, allowing for a comprehensive exploration of the relationship between the immune cell landscape and the progressive occlusion of pulmonary arteries in both hereditary and idiopathic PAH. Through meticulous mapping of microenvironmental complexities in immune infiltration, the researchers identified specific immune cell subsets linked to vascular pathology.
Their findings underscore the pivotal role of monocyte derived-dendritic cells (mo-DCs) in vascular remodeling across PAH subtypes. Furthermore, they identified neutrophils linked to heightened immune activation surrounding the pulmonary arteries, particularly prominent in severe hereditary PAH cases. This discovery opens up new avenues for potential therapeutic interventions targeting mo-DC activation and neutrophil-mediated inflammation, offering hope for managing PAH and halting disease progression.
Additional Stanford Cardiovascular Institute-affiliated investigators who contributed to this study include Aiqin Cao, Erin F. Mccaffrey, Toshie Saito, Noah F. Greenwald, Mark Nicolls, Trevor Bruce, Roham T. Zamanian, and Patricia Del Rosario.

Dr. Marlene Rabinovitch

Dr. Selena Ferrian

Dr. Michael Angelo