A Better Way to Predict Complex Genotypes with Limited Data
by Adrienne Mueller, PhD
October 20, 2023
Our immune system protects us from pathogens like viruses and bacteria, but it can also cause significant problems. Our immune system’s job is to recognize foreign material in our bodies and initiate processes to clear it. This is almost always desirable, but a primary case where it’s a problem is after tissue transplants. We want our bodies to respond to the new tissue as though it belongs to our own bodies, but our immune system is designed to flag new tissue as ‘foreign’ and activate processes to remove it. One family of genes that is especially important for the immune system’s identification of tissue as host or foreign is the human leukocyte antigen (HLA) locus. The HLA genetic locus is the most polymorphic region of the human genome and it encodes the HLA proteins responsible for presenting antigens (pieces of foreign material) that would trigger an immune response.
The HLA locus is extremely diverse – which helps us identify, present, and respond to a wide range of pathogens. The variability of alleles at the HLA locus is also one of the challenges we face in trying to reduce immune responses to new tissue in transplants. The expression of HLA genes is dynamically regulated and the expression of individual alleles – different versions of the gene – can also be highly variable. For example, the expression of a specific allele can change over time. Previous studies have shown that at any given point in time, most cells are expressing either one allele or the other of the two copies present on our pair of chromosomes. However, because the HLA genes are so extremely variable, it has been challenging to compare the result of HLA expression in new single cell sequencing studies to the large sets of publicly-available sequencing data. This challenge is exacerbated by the fact that individual cells used in single cell sequencing studies often don’t express HLA genes at a high enough level to allow genotyping.
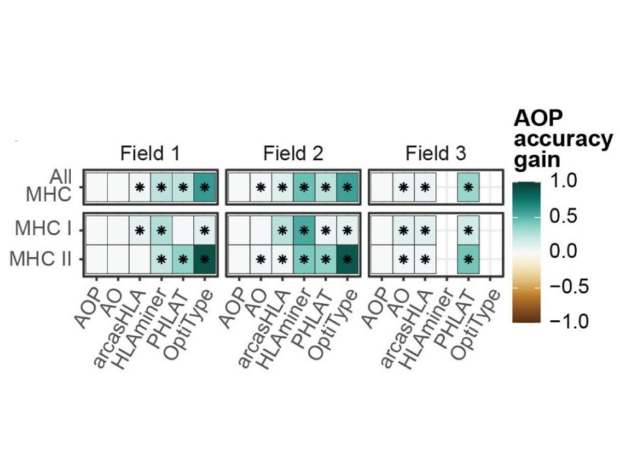
Difference in mean accuracy between the study’s composite prediction method (AOP) and different alternative genotyping methods across loci classes. *s denote p-values of less than 0.05.
Recently, a team of Stanford researchers led by Ben Solomon, MD, PhD, and Purvesh Khatri, PhD developed a new tool to overcome this problem. As reported in Frontiers in Immunology, the investigators showed that data from single cell RNA sequencing studies can be pooled into ‘pseudobulk’ sequence files that can then be used to accurately predict HLA genotypes. They compared the accuracy of predictions from five computational HLA genotyping tools of single cell RNA sequencing data to a gold standard molecular genotyping tool, and then they used a composite of those tools to try and generate a maximally accurate prediction tool.
They showed that even if individual tools produced inaccurate genotypes; combining the data from several tools can still lead to an accurate HLA genotype prediction. This new tool will help ensure that precious single cell RNA sequencing data can be utilized more easily, and more informatively, especially for complex and variable genetic loci like HLA. Additionally, this study has laid the groundwork for future experiments to better understand the HLA locus, so we can develop better systems for avoiding tissue rejection in organ transplants.
Stanford Cardiovascular-Institute affiliated author Hong Zheng, PhD also contributed to this study.

Ben Solomon, MD, PhD

Purvesh Khatri, PhD