How A Former Retrovirus Contributes to Pulmonary Arterial Hypertension
by Adrienne Mueller, PhD
August 25, 2021
Overworked Hearts
Pulmonary arterial hypertension (PAH) is a chronic disorder that progressively worsens over time and can eventually lead to death. PAH is caused by narrowing of the arteries that supply blood to the lungs. Having narrower arteries forces the heart to work extremely hard to pump more blood to the lungs and supply more oxygen to our bloodstream. Over time, the heart muscle tires and eventually fails, resulting in premature death. The vessel narrowing seen in PAH is partly caused by the abnormal expansion of the cells that make up the vessel walls. One cause for this abnormal expansion of cells is the transformation of a specific lung blood vessel cell type - endothelial cells - into a different cell type. The loss of endothelial cells triggers inflammation, further exacerbating PAH. An important question for treating PAH is therefore, what triggers the transformation of endothelial cells?
Retroviruses in Our Immune Cells
Some viruses have been embedded in our DNA for so long that they are now an integral part of our genome, producing proteins that our cells have incorporated into their regular functioning. One such protein is HERV-K dUTPase. The lab of Marlene Rabinovitch, MD has previously shown that HERV-K dUTPase levels are higher in immune cells of patients with PAH. Because PAH is caused by inflammation, an immune response, there is a strong possibility that the increase in the levels of these formerly-viral proteins are contributing to PAH. The Rabinovitch lab has also previously shown that rats treated with HERV-K dUTPase develop pulmonary hypertension - demonstrating how HERV-K dUTPase may directly contribute to PAH symptoms.
Two Pathways to a Problem
In a study recently reported in the journal JCI Insight, Shoichiro Otsuki, MD, PhD et al followed up on the Rabinovitch lab’s previous work to elucidate the specific mechanism by which HERV-K dUTPase affects PAH through immune cells. Otsuki et al showed that when monocytes, a particular type of immune cell, have an excess of HERV-K dUTPase, they shed the protein via small packets called extracellular vesicles. The investigators further showed that shed HERV-K dUTPase, which mediates gene expression in lung endothelial cells, initiates signaling cascades that trigger the transformation of endothelial cells into a different cell type.
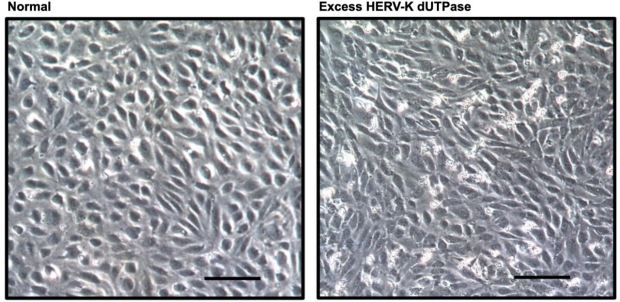
Compared to normal cells (left), excess HERV-K UTPase (right) causes lung endothelial cells to change morphology and lose endothelial cell properties becoming more like smooth muscle cells.
The investigators showed that HERV-K dUTPase specifically activates two complementary signaling pathways that each trigger the abnormal transformation of endothelial cells. This transition provokes inflammation and is therefore likely to ultimately cause the inflammatory responses underlying PAH. Otsuki et al have not only demonstrated a mechanism underlying PAH, but also pointed the way to the development of novel therapies for PAH that target HERV-K dUTPase pathways.
Additional Stanford Cardiovascular Institute-affiliated investigators who contributed to this study include Toshie Saito, Shalina Taylor, Dan Li, Jan-Renier Moonen, David P Marciano, Rebecca L Harper, Aiqin Cao, and Lingli Wang.

Shoichiro Otsuki, MD

Marlene Rabinovitch, MD