Calcineurin Bound — A New Mechanism
to Help Explain Heart Failure
By Adrienne Mueller, PhD and Amanda Chase, PhD
August 21, 2020
The Calcium Conundrum
Chronic stress can ultimately lead to heart failure. One explanation for this is that stress-related signals trigger an unwarranted and pathological growth of our heart cells. These pathologically enlarged (hypertrophic) heart cells do not function well, which causes the heart to pump blood poorly, eventually resulting in heart failure.
A protein that plays a central role in this pathological heart cell enlargement is the Ca2+-activated protein Calcineurin— specifically Calcineurin Aβ. Once activated by Ca2+, Calcineurin Aβ causes a cascade of signals that lead to hypertrophy. What is perplexing about this is that heart cells are flooded with calcium all the time—with every heartbeat. In fact, Calcineurin Aβ should be promoting heart cell hypertrophy as soon as we have a pulse, and yet that is not the case. In seeking to understand why this is so, a group of Stanford Cardiovascular Institute-affiliated researchers, including co-first author Jinliang Li, PhD, and senior author Michael Kapiloff, MD, PhD, uncovered not only an explanation for this conundrum but also a mechanism for how stress could lead to Calcineurin-related heart failure and a potential means of preventing it.
The Advantages of Compartmentalization
In their recently published paper in Circulation, Li & Li et al demonstrate that in heart cells, Calcineurin Aβ is restricted to cellular "compartments" that shield it from the large Ca2+ transients that occur when our heart beats. What’s keeping Calcineurin Aβ in these compartments? A molecular tether: in this case a scaffold protein called CIP4, that specifically anchors Calcineurin Aβ within CIP4 compartments and prevents it from being exposed to Ca2+ that floods the rest of the cell. Using state of the art imaging techniques, the researchers showed that when they simulated disease-related stress, Ca2+ and Calcineurin Aβ activity specifically increased in CIP4 compartments of cells but not the cells as a whole. Chronic stress may therefore allow Ca2+ to enter CIP4 compartments, activating Calcineurin Aβ, and leading to pathological heart cell remodeling and heart failure.
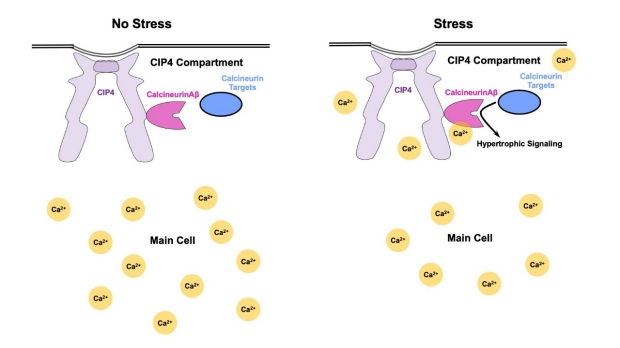
Our heart cells use molecular compartments to keep Ca2+ away from Calcineurin Aβ, which would normally elicit a cascade of signals resulting in hypertrophy. When the system is pathologically stressed, Ca2+ may enter these compartments, activating Calcineurin Aβ, which then leads to hypertrophy.
Holding Back Heart Failure
Li & Li et al not only showed that Calcineurin Aβ is localized to these CIP4 compartments, but also that stopping the tethering of Calcineurin Aβ to CIP4 reduced pathological heart cell enlargement both in cultured heart cells and in living mice. Indeed, their intervention reduced the detrimental cardiac remodeling that heart cells undergo after simulated heart failure.
Li & Li et al discovered a structural model that explains why our heart beats do not cause our hearts to grow pathologically large. Our heart cells use molecular compartments to keep Ca2+ away from Calcineurin Aβ, which would normally elicit a cascade of signals resulting in hypertrophy. When the system is pathologically stressed, Ca2+ may break into these compartments, activating Calcineurin Aβ, which then leads to hypertrophy. Li & Li et al’s results suggest that therapies that disrupt Calcineurin Aβ CIP4-tethering are a promising avenue to help prevent heart failure.
Other Stanford Cardiovascular Institute-affiliated authors include Yang Li, Qian Yu, and Hrishikesh Thakur.

Dr. Jinliang Li

Dr. Michael Kapiloff